Study Finds FDA 510(k) Approval Loophole Linked to Class I Recalls for Medical Devices

A loophole in an FDA approval pipeline means medical devices with serious safety shortcomings can still serve as blueprints for new products.
Several 510(k) devices subject to Class I recalls in the States use predicates which themselves have a known history of Class I recalls.
Nearly all medical devices in the US progress to market under the 510(k) pathway, which uses previously authorised devices – or predicates – to support new authorisations.
Now, research has suggested a cause for rising trend in devices approved though this pathway being subject to a Class 1 recall: the FDA’s most serious label for a faulty medical device.
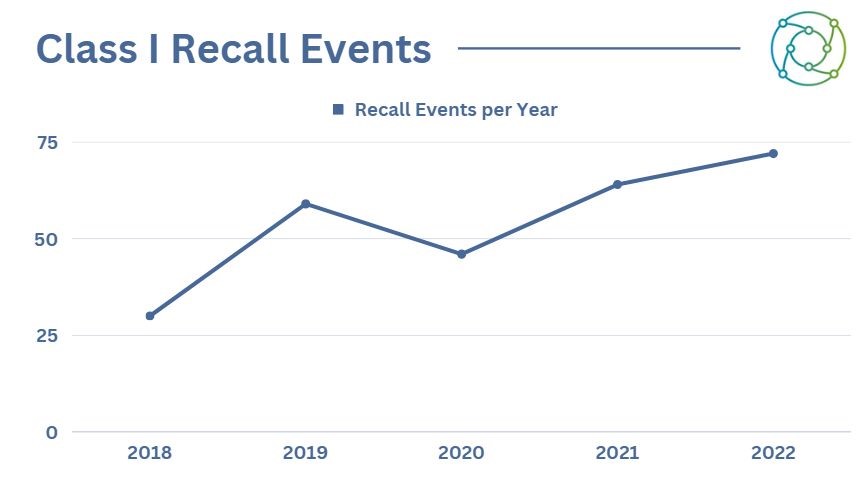
2022 saw more Class I recall events, continuing a slight rising trend that has become more established over the past five years.
Examples of medical devices that have previously been recalled include chronic dialysis catheters, ventilators, and defibrillators.
Researchers from Harvard, Yale and the University of California looked at the composition of devices recalled between 2017 and 2021 (Kadakia, K.T. et al., 2023).
Through tracing the medical device genealogy for every device issued with a Class 1 recall in this period, they found devices were more likely to be recalled if the FDA authorisation was based on devices that had themselves previously been recalled.
This likelihood was compounded for future generations of devices cleared through the same pathway.
Of 127 devices subject to a Class I recall between 2017 and 2021, nearly half (44%) had been authorised based on predicates with their own Class I recalls.
Overall, devices were 6.4 times more likely to be subject to a Class I recall if they received clearance based on a previously recalled ancestor.
A Flaw in the 510(k) Approval Pipeline
The FDA’s 510(k) approval program receives the majority of device submissions across the US each year.
It was devised as a quicker alternative to the full approval process: approval is granted if a device maker can prove their new device is ‘substantially equivalent’ to that of a previously cleared device as being both safe and effective.
Research analysis found that current regulations permitted manufacturers to use devices which had been subject to a Class I recall as ‘predicates for new devices’.
Even after a device has been associated with a ‘heightened risk of injury or death’, it may still be used as a blueprint for the subsequent development of new products which are then submitted to the FDA for clearance.
Related Articles
- Medical cannabis manufacturer Celadon Pharmaceuticals awarded GMP registration
- Subcutaneous injection: getting drugs where they need to be in the body
Dr Harlan Krumholz, Harold H. Hines Jr. Professor of Medicine (Cardiology) and director of the Yale Center for Outcomes Research and Evaluation (CORE) at Yale School of Medicine (YSM), was quoted by Yale News as saying this was “not an FDA issue, but [it] is about the law that governs FDA actions.”
To break this cycle and prevent the use of predicates which may continue to provide a problematic genealogy for future medical devices, safeguards for the 510(k) approval pipeline have been recommended.
Congressional action is likely required to improve medical device safety and reduce the risk of future recalls.
Get your weekly dose of industry news and announcements here, or head over to our Formulation portal to catch up with the latest advances in manufacture and therapeutic delivery.
Kadakia, K.T. et al. (2023) “Use of recalled devices in new device authorizations under the US Food and Drug Administration’s 510(k) pathway and risk of subsequent recalls,” JAMA, 329(2), p. 136.
Related Resources