










Industry Insights Into the FDA's Plans for Revised Gene Therapy Approval Approaches

The FDA has recently announced a sweeping range of overhauls and alterations it intends to put in place to accelerate its cell and gene therapy approval pipeline.
These include the use of adaptive studies, novel endpoints, and new approaches to statistical analysis.
Susan Stewart is Chief Regulatory Officer at Candel Therapeutics, and has a comprehensive background in biopharmaceutical regulatory affairs spanning more than 30 years.
“The FDA are putting a lot more power into these fast-track and breakthrough therapy designations,” she explained in an exclusive Q&A with Oxford Global.
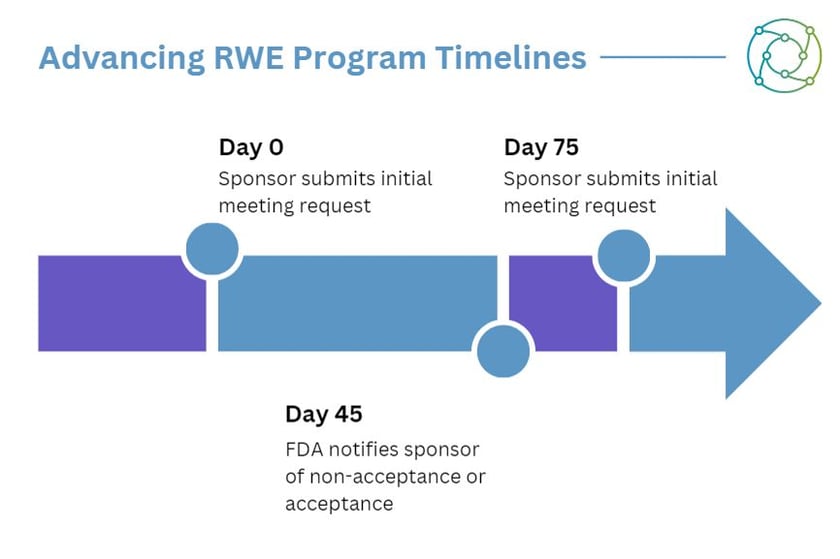
This will involve enhancing the drug development tools qualification pathway for biomarkers, use of real world evidence in efficacy assessments, increasing staff capacity and training, among a slew of initiatives to enable efficient product development and review.
How Will RWE be Integrated into Cell and Gene Therapy Trials?
One key alteration to the gene therapy approval pipeline is the interpolation of real-world evidence (RWE) into trials.
The FDA is constructing a methodology to improve its understanding of robustness evaluations used to address the consistency of RWE with respect to study design, analysis, and variable measurement.
Enabling the use of RWE to assess the efficacy of therapeutics could help to hasten approvals for products with accelerated clinical development.
Also of interest is the regulator's CMC development and readiness pilot program, which is just starting to receive applications.
“If your project gets a designation as a breakthrough therapy program, the FDA will literally hold hands with you to try to coordinate in many areas,” said Stewart.
“Right now, the best we've seen the FDA consider real-world evidence is to support safety claims.”
As the regulator continues to look for approaches that will confer advantages over existing therapies, this may change.
Will the FDA Accept Alternatives to Animal Models in Product Validation?
Another major announcement that came in December 2022 was the alternative methods program.
The USA's President Joe Biden lifted the requirement that pharmaceutical companies test new drugs on animals prior to human trials.
The move is intended to reduce animal testing through the development of qualified alternative methods, such as computer modelling and organ-on-chip approaches.
However, Stewart cautioned that the FDA could still require data validated through animal models until these alternative approaches are more established in the gene therapy approval pipeline.
“Everyone sees it's important that gene therapies are available to patients who need them.”
“The FDA could still require it, and my feeling is that this will still be a bit of a long road for getting to some equilibrium about how that animal data could be used,” she said.
“We have to have some basis for having chosen a dose or claimed safety or feasibility of a treatment before testing in a person.”
Some additional measures announced in the FDA overhaul include the launch of the Cell and Gene Therapy Access Model, which aims to increase the availability of new therapies once they come onto the market.
“The encouraging part is that everyone sees that it's important that gene therapies are available for patients who need them,” added Stewart.
Get your weekly dose of industry news and announcements here, or head over to our Cell portal to catch up with the latest advances in cellular therapies.
Related Resources

