




Sparsely Connected Autoencoders: A Multi-Purpose Tool for Single Cell Omics Analysis
Presented by: Raffaele Calogero, Associate Professor of Molecular Biology at the University of Turin
Edited by: Ben Norris
In single cell omics analysis, different cell types are not easily quantifiable without the removal of experimental noise. As such, a reduction in dimensionality is crucial for the visualisation and interpretation of single-cell sequencing data. Raffaele Calogero, Associate Professor of Molecular Biology at the University of Turin, has recently developed a new class of autoencoders: artificial neural networks used to address a biologically driven data reduction in single cell RNA-seq data. In his research, Sparsely Connected Autoencoders represent the bioinformatics version of a Swiss Army Knife for the extraction of hidden bioinformatics from single-cell omics data.
Clarification of Cellular Structure: De-Noising Data
“Bioinformatics should be used on the basis of the biology that you are studying,” opened Raffaele Calogero at Oxford Global’s Spatial Biology Europe 2022 conference. He highlighted the importance of dimensional reduction in single-cell analysis, adding that single cells are, in general, “very noisy and highly dimensional.” This necessitates the use of Sparsely Connected Autoencoders (SCAs) to depict biologically interesting features from single-cell omics data.
“You get, as input, your noisy single-cell data,” Calogero explained. “Everything is compressed and then deconvoluted in order to remove experimental noise and enhance cells biological characteristics.” In doing so, this enables the reconstruction of the overall picture of cells organisation in a tissue. This reduction in dimensionality is achieved through the projection of high-dimensional data into low-dimensional spaces to visualise the cluster structures and difference in developmental trajectory.
Sparsely Connected Autoencoders: Deep Learning Approaches
The solution Calogero and his team arrived at was a deep-learning approach utilising SCAs, which he described as being ‘very powerful’. The autoencoder is an unsupervised artificial neural network designed to reduce data dimensions by learning how to ignore both noise and anomalies in the data. “It’s a relatively simple neural network,” continued Calogero, “characterised by input and output data representing general gene expression.”
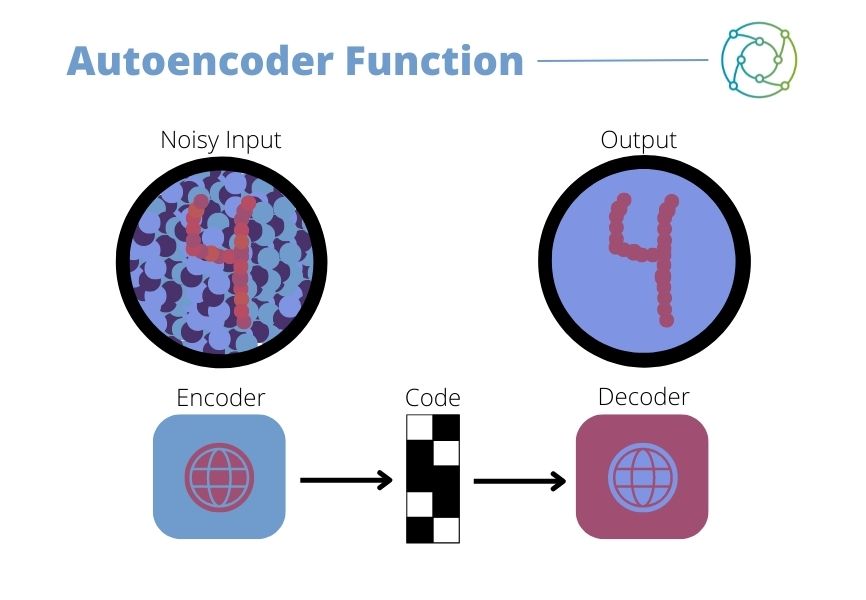
The autoencoder is an unsupervised artificial neural network, designed to reduce data dimensions by learning how to ignore noise and anomalies in data. It compresses and encodes data before reconstructing it from the reduced encoded representation. In doing so, it produces an output that is as close as possible to the original input (seen in Figure 1). The autoencoder utilises a single-later with sparse connections to attain a value for biological features and not the simple gene expression. “This approach has the advantage of being easily extended to any gene set designed around some biological feature, such as functional networks or gene regulatory elements such as transcription factors,” Calogero added.
A study undertaken by Calogero and his team attempted to reconstruct breast cancer data using a spatial transcriptomic dataset. “When you look at clustering on SCA processed data, you realise that some clusters have intracluster heterogeneity which cannon be easily depicted using the simple gene expression,” he said.
Depicting Hidden Cell Type Characteristics
Calogero then moved on to discussing the Colon Immune Atlas. The colon, as a barrier tissue, represents a unique immune environment where immune cells display tolerance towards diverse communities of microbes collectively known as the microbiome. The Colon Schematic Study – an investigation of single-cell transcriptomes for 41,650 cells isolated from the caecum – revealed differences between the immune cells in different parts of the colon.
- Monitoring Immune Suppression in the Tumour Microenvironment
- Long and Short Read Sequencing: Revolutions in Genomic Data
- Event Showcase: Sequencing the Future at Spatial Biology Europe
As Calogero explained, the immune cell types in the Colon Immune Atlas were annotated using a limited number of markers and the separation among the immune compartments was not easily depictable using conventional single cell transcription profiling. By representing the data using SCAs based on transcription factors, Calogero found that the separation among cell types was more evident along different immunological cell origin, highlighting the differences based on cell specific transcription control.” Additionally, cell type annotation could be refined using SCA transformation.
“In an oncological setting, the idea was to try to investigate if we could identify differences in, for example, a population that seems to be relatively homogenous,” said Calogero. Analysis of this type of dataset involved the counting of matrices produced by a mixture of five human lung adenocarcinoma cell lines. “The interesting part is that, after SCA deconvolution by mean of transforming expression data in cytoband expression, differences within a relatively homogenous cell line can be detected.”
Notes for Future Applications of Sparsely Connected Autoencoders
Cell subpopulation can be depicted simply using the gene level information. However, when expression data is aggregated on the basis of some biological feature or genome structure characteristic – for example, transcription factors or cytobands – hidden subtle cell population structures start to become apparent. “The work we are now doing is collecting data on how the open chromatin is changing in cell differentiation,” Calogero said.
The focus for future projects will involve building a new model where different biological features are characterised by their interactions with one another. For example, kinases, transcription factors, miRNAs, and chromatin organisation, where kinases drugs can be tested in silico with a view to identifying key elements involved in their functionalities. “From a technical point of view, it’s very difficult to generate these kinds of models,” he said. However, the benefits for healthcare studies are manifold, with huge implications for the future of single-cell omics analysis.
“The ability of the autoencoder to retain only the important part of a signal can help in discriminating true differences in cell populations.”
SCAs offer the opportunity to transform data in a controlled way to enable the comprehension of hidden biological information in single-cell data. This information may include relations among cell populations. SCAs also provide researchers with a simplified means of inspecting functional relationships among regulatory elements as transcription factors, or miRNAs. “The peculiar ability of the autoencoder to retain only the important part of a signal can help in discriminating between true differences among cell populations and clustering overfitting,” concluded Calogero.
Visit our Omics portal to read more about the latest advances in single-cell analysis and other breakthroughs in genomic data imaging. If you’d like to register your interest in our upcoming Spatial Biology US: In-Person event, click here.
Related Resources
