




Exploring Advanced Multiplex Assays

Our April omics discussion group brought together key opinion leaders in DPCR, including representatives from AstraZeneca, Biogen, University of Florence, and University College London. The group focused on novel applications of DPCR technology with insights into advanced multiplex assays and DNA-based deconvolution of the tumour microenvironment.
This month’s group was led by Pieter van der Velden, at Principal Investigator at Leiden University Medical Centre. Pieter works in the department of Ophthalmology, specialising in the field of uveal melanoma genetics. By using the most recent technical advances in DDPCR, he measures genetic and cellular heterogeneity. Pieter is also part of UM Cure, a European consortium of preclinical researchers that aims to provide treatment for metastatic uveal melanoma.
Multiplex Assays & T-Cell Counting
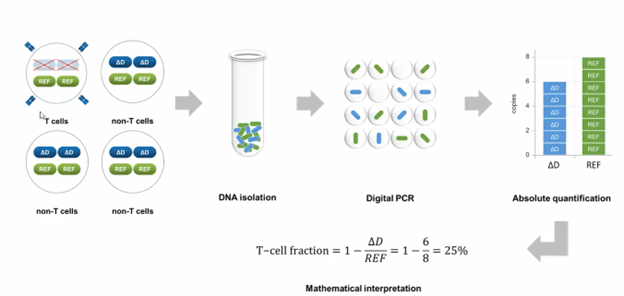
Pieter opened the discussion with an explanation of the T-Cell quantification method his team developed for Digital PCR. He explains that “it is based on a marker in T-Cell receptors. This can be the TCR delta or TCR beta, that is lost in all T cells. If you have, like in the example (picture above), four cells, and one is a T cell, all cells have two reference genes and two delta D alleles except T cells. The T cell loses, specifically, this marker. Now, if we isolate DNA, put it in droplets, and we start counting them, we will find delta d in the six droplets, and we find eight droplets positive for the reference. So, we have four cells, and there's only six delta D alleles that makes 25% T cells.”
This method has been described in several papers. To gain further insight, multiplex T/B cell counting can be used to create an accurate marker for quantitative immune cell analysis. Pieter explains that “It's all based on negative counting with the specific markers that are always lost in B and T cells upon maturation.”
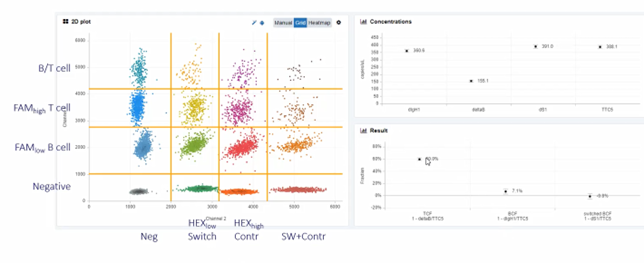
Deconvolution of the Tumour Microenvironment
The heterogeneous cell types of the tumour microenvironment are pivotal in predicting cancer progression and treatment response. One of Pieters's team’s areas of interest is uveal melanoma. Pieter shared his team’s findings, saying, “we found that most advanced tumours have around 75% tumour cells. Early tumours have a much higher tumour cell percentage. If you looked at the expression profile, you could clearly see the difference. The gene expression also gives an indication of what's present. So, we also found the markers for CD8 and CD3 in this expression profile that classifies highest progression tumours.”
CD3 and CD8 T cells are increasingly being investigated to better understand their links to inflammation, immune disease and cancer. Pieter clarifies his position; “you can, of course, use gene expression profile software to calculate fractions, but the basis of these tools is rather weak correlations between gene expression and real cell fractions. In order to study this in more detail, we believe that DNA is a much better parameter for counting cells. Based on the counts that we did on those tumour cells; we reanalysed the expression profile and examined correlation to T cells. We came up with a whole list of genes that are highly correlated both negatively and positively with T cell numbers that explain the inflamed phenotype.”
DNA Methylation
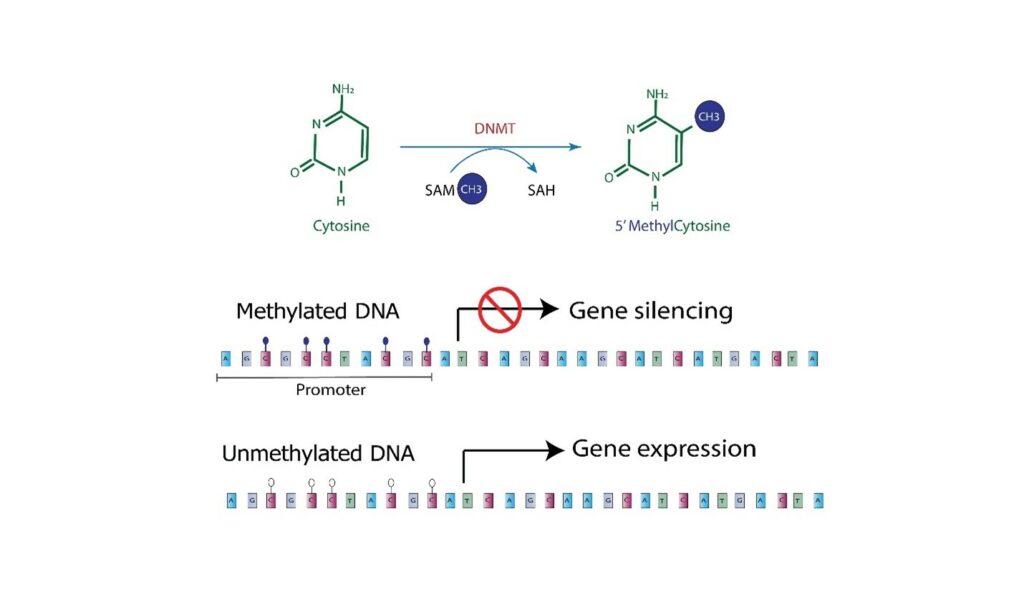
The previous studies involved a bisulfite conversion; however, this can lead to issues with loss of materials or incomplete conversion. DNA methylation is an epigenetic mechanism that occurs by the addition of a methyl (CH3) group to DNA, thereby often modifying the function of the genes and affecting gene expression.
Pieter explains his teams use of restriction enzymes to determine the methylation status of DNA; “we switched to methylation sensitive restriction enzymes to determine the methylation status of DNA. in this approach, we first perform a baseline experiment, to determine the copy number of your target in the tissue that you analyse. Next, we do an experiment in which we add the methylation sensitive restriction enzyme. If the methylated amplicon is not digested and can be used to generate a signal droplet, whereas an unmethylated target is fragmented by the restriction enzyme.” This method sparked significant interest in the group, leading to several key questions about the process.
DNA Methylation and Restriction Enzymes: Key Questions:
Within your target, do you always find that the CPGs are either 100% methylated or 100% non-methylated?
No, in the example I spoke about today there 6%. methylation. And that's what's confirmed, 94% was unmethylated. Every droplet that that remains present, represents a methylated copy. We do mixtures, and the pattern is never homogeneous. Our team prefers to design based on the NGS data, for example, if you do bisulfite sequencing of a locus, we know what CPGs are informative. We have a software tool to select specifically the CPGs for our specific cells.
How did you choose your restriction enzyme?
We have NGS data and then made the profiles. We have a favorite enzyme, BstUI which we preferentially use, but we have had to use other enzymes. It's about information; how discriminating are the methylation profiles, the specific CPGs. It’s important to be aware of this information when you start developing markers and of course the proof is in the testing of the markers with digital PCR.
What was the decision process that led to this enzyme-based method? you mentioned the bisulfite conversion. Have you tried the Enzymatic Methyl-seq Conversion modules?
With bisulfite, we were losing material creating and the risk of incomplete bisulfite conversion, which can lead to a skewed conclusion. We found we could conclude more confidently using restriction enzymes. The enzyme we use, BstUI, works well at high temperature and in the reaction buffer.
Final Thoughts & Conclusion
At Oxford Global, we couldn’t be more pleased with the turnout and feedback from this discussion. This meeting provided the perfect setting for exchanging ideas, sharing innovations, and discussing the ever-evolving DPCR landscape. For more on PCR, our NextGen Omics UK event will feature two PCR tracks with presentations, workshops and more.

