




Developing Meaningful Assays: Balancing Biological Significance and QC-Compatibility
Presented By: Katrina Adlerz, Scientist at The University of Texas MD Anderson Cancer Center
Transcribed By: Tia Byer
The Cell Therapy Manufacturing Facility is located on the main campus of the University of Texas MD Anderson Cancer Center ND and is responsible for bringing together both development and Good Manufacturing Practice (GMP) in autologous cell therapies. In particular, the group's main focus is to translate products from preclinical stages to early phase clinical trials with the goal of achieving biological application for commercial therapies.
Biologically Meaningful Assays and FDA Guidance
The Cell Therapy Manufacturing Facility group uses established safety testing methods during all phases of product development. Key strategies include sterility, endotoxin, and mycoplasma testing. Similarly, testing for dose is required to determine biological meaningfulness.
Potency assays are the most biologically meaningful assay. Potency is defined by the Code of Federal Regulations as “the specific ability or capacity of the product, as indicated by appropriate laboratory tests or by adequately controlled clinical data obtained through the administration of the product in the manner intended, to effect a given result.”
Potency assays must be validated by the Biologics Licence Application, and the FDA lays out a Progressive Policy Assay Implementation strategy in their guidance (see figure. 1). They instruct that “as clinical study progresses and product knowledge increases, you should develop and implement improved potency measurement(s) that quantitatively assesses relevant biologic product attribute(s).”
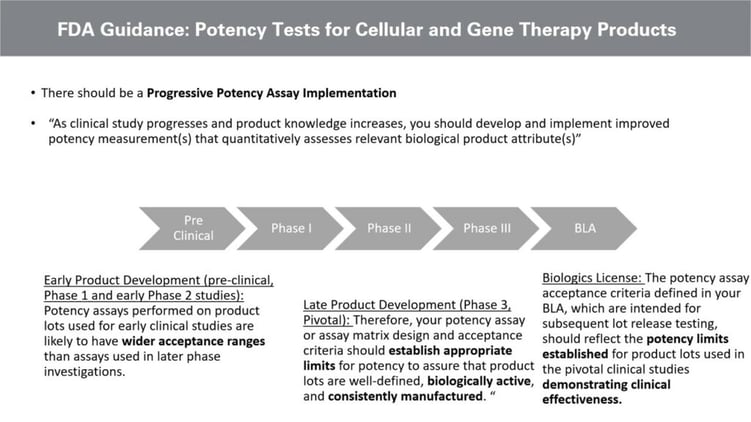
Whilst there is a broader acceptance criterion in the early development stages, later potency assay development requires researchers to establish appropriate limits to ensure the product is biologically active and consistently manufactured.
The Necessity of Validation in Early Analytical Development
An assay design should be considered as early in development as possible to ensure validation compatibility. This includes selecting reagents that are controlled by the supplier and selecting GMP grade reagents where applicable. Operator independence, precision, accuracy, and some of the other validation parameters listed in the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines should all be considered during assay design.
The goal of an assay is to be both accurate and precise...an assay that is neither accurate nor precise would not be acceptable.
In addition to this, there must be a balance between trying to produce biological significance whilst implementing validation compatibility. The goal of an assay is to be both accurate and precise. An assay that is neither accurate nor precise would not be acceptable. However, it may be possible that a precise but slightly inaccurate assay is more suitable for a release assay than an accurate but imprecise assay.
Assay Specifications: Types of Acceptance Criteria
Another essential consideration for assay design is the specification. A quantitative specification is more appropriate than a qualitative specification for release assays. There are a number of ways to define a quantity of specification, with one being to provide acceptable ranges (see figure. 2). Alternative strategies include identifying bins greater than or less than and comparing reference material.

Currently, there remains a lack of standard reference material in the field. Efforts are being made with standards coordinating bodies such as the National Institute of Standards and Technology and some working groups such as IS Biotech group working on lentiviral reference material. However, as of now, reference material is better suited as an assay control or to define assay validity versus assay specification. As such, the Cell Therapy Manufacturing Facility has been working on one with a binary readout, with specification summarises the readouts of a population-based metric.
Assay Design Example: Tumour Infiltrating Lymphocytes (TIL)
Another assay the group is currently developing is the TIL, which is a simplified schematic of the autologous T Cell therapy and applies the beforementioned general principles of assay design. For this assay, tumour cells are taken from the patient and put into a culture to expand the tumour infiltrating lymphocytes. A rapid expansion process called Rep then takes place, where billions of TIL are generated to then be infused back into the patient.
- Inciting Rapid Standardisation in Cell and Gene Therapy Manufacturing
- What are the Foremost Challenges and Opportunities in Viral Vector-Based Gene Therapy
- Find out how to Overcome the Key Challenges of Gene Therapy Commercialisation
Cytokine secretion is one measure of biological activity. TIL offers a unique challenge in defining biological activity compared to other cell therapies like CAR T. With CAR T-cell therapies, for example, the specificity to the cancer cell is engineered, which means a CD19-CAR T acts against CD19 positive cancer cells. Therefore, CD19 positive cancer cells can be used as a target cell that stimulates cytokine secretion in a potency assay.
TIL, on the other hand, does not possess a specificity that is engineered-based. Instead, its specificity is tumour-based, whereby the TIL has been isolated from the tumour and will, therefore, most likely react against differing antigens. This process makes the therapy effective but the assay design challenging.
Assay Design Example: Cytokine Secretion Assay Design
With a general assay design for cytokine secretion, activating cells to secrete cytokines such as Interferon-gamma and Tumour Necrosis Factor alpha is necessary. A supernatant method, such as the plate-based ELISA, can then be used to analyse the activity. Validation and compatibility should be considered upon selecting the supernatant analysis method, along with linearity and accuracy precision. The variability of the assay is most significant at the activation stage (see figure. 3).
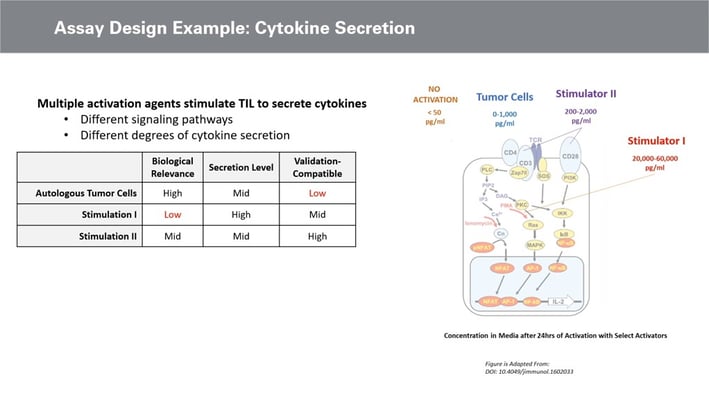
Therefore, the biological activation of cytokine is a key decision in assay design. Optimal methods to achieve cell activation include using autologous tumour cells or autologous tumour digest. The biological relevance of using these tumour cells to stimulate the TIL is high, whilst the validation compatibility is very low and difficult to determine.
Conclusion and Summary
For assay design, artificial stimulation using activators such as CD3 or CD28 offers comparably higher validation compatibility. Biological relevance is also within the same range as the tumour cells regarding cytokine release. Furthermore, artificial stimulators possess an optimal balance between biological meaningfulness and QC validation compatibility.
Whilst it is possible to determine the biological activity of cells during the early stages of development, validation is not possible upon entering the commercial phase. Biological meaningfulness and validation compatibility are likewise important. Therefore, validation considerations for cell and gene therapy development must be implemented as early as possible to ensure biological license application.
Want to find out more about the latest cell therapy news? Register now for Oxford Global‘s Cell UK: In-Person event to advance your understanding of cell-based products to ensure clinical and commercial success.
Related Resources

