




De Novo Variants: Genomic Sequencing and Problem Solving for Future Treatments

Presented by Hywel Williams, Senior Lecturer at Cardiff University
Transcribed by Ben Norris
The intricacies of the human genome have been unravelled by the completion of the Human Genome Project, and the speed at which it can be sequenced has gone from ten years to tens of genomes in a day. However, bottlenecks remain in the context of genome sequencing and data interpretation. Hywel Williams, Senior Lecturer at Cardiff University, explained to the audience at our NextGen Omics UK: In-Person Event last November that there may be a way around the delays inherent with human and laboratory analysis of genomes.
Monitoring for De Novo Variants in Genomic Data
As anyone caught in traffic during a lane closure will know, bottlenecks can massively slow down progress – in the context of the morning commute, or equally in the context of genomic sequencing. As Hywel Williams explained, recent advances in the speed of sequencing have brought with them new speedbumps on the road to patient diagnosis and treatment. “We’re sequencing more patients in the clinic, and therefore we’re producing more data, and we’re hitting bottlenecks.” He continued that if it was possible to increase the speed taken to reach a diagnosis, it would facilitate an increase in the diagnostic rate.
Williams added that this will allow more time to inspect the ‘tricky’ patients: “those patients that may have novel variants that take a bit more time to look at than they normally would.” De novo gene variants are genetic alterations that are present for the first time in one family member because of a mutation in a parent germ cell. Williams and his research team at Cardiff have placed a particular research focus on de novo variants as a key area to address in the resolving of bottlenecks in genetic therapy. The bottleneck mentioned previously comes from the clinical interpretation phase associated with making a diagnosis: pathologists and oncologists must evaluate the significance of all potentially clinically actionable events.
Constrained Coding Region (CCR) Data
“If we think about next-gen sequencing and data bottlenecks, sequencing a whole genome was something that took a long time,” Williams continued. “I can remember the days when it could easily take over a week to analyse a single human genome. Now we’ve improved algorithms, we can use bespoke genomic hardware to do that in 30 minutes.” However, this leaves Williams and his team with around three million variants in the dataset, which is “a lot to go through.” Fortunately, research assets such as the gnomAD database enable researchers to rapidly trawl through the data and remove variants that are not interesting or relevant, since they are known to be too common in the population to be relevant to the patient’s phenotype.
"It could easily take over a week to analyse a single human genome... now we can use bespoke genomic hardware to do that in 30 minutes."
Williams was inspired by the approach to genomic analysis in a 2019 paper by James Havrilla from Aaron Quinlan’s Group in Utah. “What they effectively did was take the gnomAD database and look at functional protein altering variants,” he explained. This was done with the knowledge that the vast majority of known pathogenic variants affect the coding sequence of proteins: by analysing the sequence, functional variants could be identified in the healthy database.
Williams continued: “They worked out that on average every seven base pairs along the coding sequence you will see a functional protein variant.” The red flag for identification came with certain regions of coding genome which did not exhibit a functional variant for 50 or even 100-plus base pairs. “The reason that there are no variants within these regions is that if you do have a functional variant, it’s likely to cause a rare disease,” added Williams. “And if you have a rare disease, you’re not going to be in a gnomAD database.”
Sequencing and the 100,000 Genomes Project
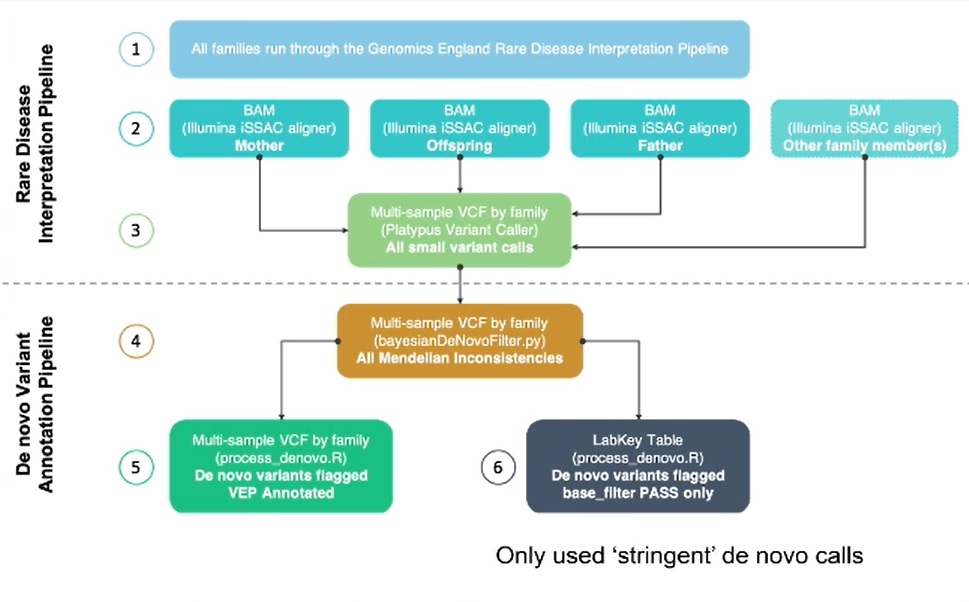
The dataset used by Williams and his team for this analysis was the 100,000 Genomes Project (100KGP); an initiative led by Genomics England that performed whole genome sequencing on over 100,000 NHS patients and their families, affected by rare diseases or cancer (100,000 Genomes Project Pilot Investigators, 2021). All families studied in the research project were run through the Genomics England Rare Diseases Interpretation Pipeline to identify potential pathogenic variants. The clinical analysis of these is ongoing. However, for this study, Williams had diagnostic data available on over 8,000 patients (GMC Exit questionnaire). Additionally, a subset of these patients were analysed using a bespoke analysis pipeline to identify de novo variants. It was the overlap of patients in these two datasets that Williams used for his study.
- Multi-Tissue Molecular Profiling: Precision Medicine for Neurodegenerative Diseases
- Single Cell Proteomics: Sample Preparation and Tissue Selection
- ddPCR: Exploring the Progress of PCR Technology and Novel Applications
There are several challenges associated with analysing large datasets such as the 100KGP. “This was by far the most difficult part of this whole project – the data wrangling required to get to the end probably took me a good few months,” said Williams. He gave several reasons for this, one being the size of the dataset: “We’ve got 35,000 observations in the GMC Exit questionnaire and we’ve got 91,000 de novo calls. But data quality was also an issue, in the GMC Exit questionnaire, we saw many instances where a variant in the same sample was duplicated.”
Furthermore, some non-pathogenic variants were classified as pathogenic and had not been entered properly. To counteract this, all variants were individually inspected to track for accidental misinformation. The final challenge was due to the data being present in two reference genome builds (GRCh37 and GRCh38). This was overcome by uniting the two datasets using the latest build (GRCh38).
Results and Findings of Rare Genetic Disease Analysis
The end result for Williams and his team proved to be incredibly fruitful. “Once I had all the data together, I could match it to see if it was a de novo variant, intersected a CCR or both” he explained. “Just by applying a de novo filter, you can pull out 40% of your pathogenic variants.” Moreover, of the variants that intersect the top 5% most constrained CCRs, 84% are clinically classed as pathogenic and if consideration is limited to those de novo variants that also intersect these CCR regions 87% are pathogenic. Putting this in the context of whether applying these annotations can improve the time taken to identify pathogenic variants, Williams said “If we go back to our hypothesis and we look at the speed taken to reach this, it will reduce the time taken to reach a diagnosis.”
When asked about the potential applications of AI in reviewing and accelerating the data processing necessary for the investigation, Williams added there are a number of AI approaches for the classification of deleterious variants. “But as far as I’m aware, none of them utilise CCR data. And that’s something again that I’d really like to try and incorporate into one of those AI algorithms.”
Future projects for Williams and his team include a follow-up on putative pathogenic variants through the Genomics England portal. This will hopefully enable the performance of these annotations to be made on a larger dataset, which would ideally include the generation of de novo data for the entire 100KGP. “In terms of future work, I want to send these details back to genomics England. The next stage is to see if it has utility in the clinic.”
Visit our Omics Portal to read more about the latest advances in genomic analysis and research. If you'd like to register your interest for our upcoming NextGen Omics UK: In-Person event, click here.
References
Havrilla, J., Pedersen, B., Layer, R. and Quinlan, A., 2019. A map of constrained coding regions in the human genome. Nature Genetics, 51(1), pp.88-95.
100,000 Genomes Project Pilot Investigators, 2021. 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care — Preliminary Report. New England Journal of Medicine, 385(20), pp.1868-1880.

