Combination Products: Navigating Recent Regulatory Changes
The regulatory pathways for combination products are complex and have significant differences across jurisdictions; even what constitutes a 'combination product’ varies by region.
The FDA defines combination products as “therapeutic and diagnostic products comprising two or more regulated components, i.e., drug/device, biologic/device, drug/biologic, or drug/device/biologic, that are physically, chemically, or otherwise combined or mixed and produced as a single entity. The FDA defined three categories of combination product: Single entity, Cross-labeled and Co-packed.
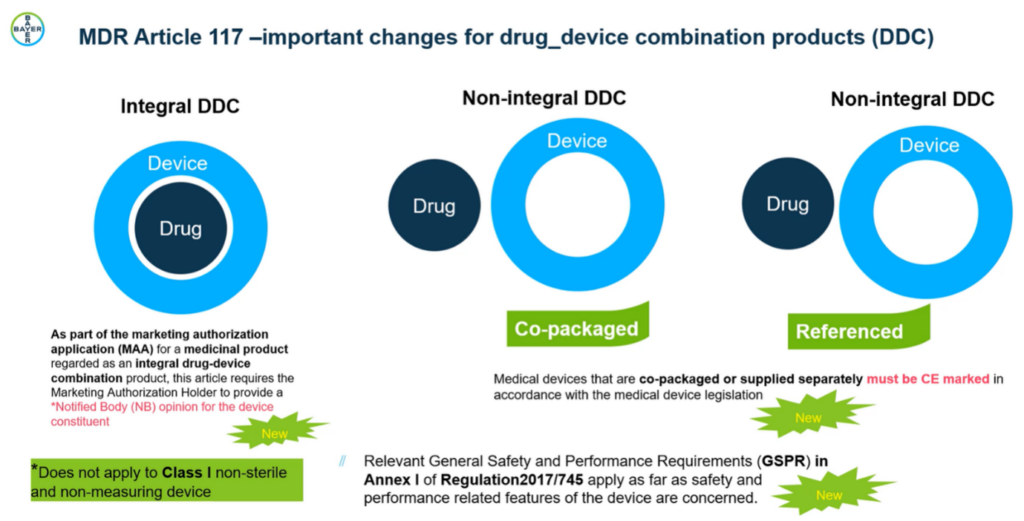
In the European Union, the Regulation for Medical Devices (Regulation (EU) 2017/745) included articles addressing devices designed and sold with a medicinal product as an integral part. These drug-device combinations are separated into two categories:
- Article 1(8): Devices incorporating as an integral part a substance that, if used separately, would be considered a medicinal product and the action of the substance is principal.
- Article 1(9): Devices intended to administer a medicinal product where they form a single integral product intended exclusively for use in the given combination which is not reusable.
Based on the Medical Device Regulations (MDR) definition, the European Medicines Agency (EMA) differentiates three types of combination products in European Union.
Integral consists of at least two constituent parts, one of which is a device, which are combined (e.g. physically, chemically) in such a manner that they form a single entity when placed on the market (equivalent to single entity by the FDA). Non-integral drug devices separate the medicinal product but are sold as a single package. Finally, referenced products are medications intended for use with a specific medical device described in the product information. These varying definitions, alongside the nebulous and rapidly nature of combination products create a challenge for novel therapeutics in this space.
Benefits of Combination Products
Despite the challenges, there are several advantages that make combination-drug products worth pursuing. Research has shown combination products to have improved therapeutic effect, enhanced efficacy, increased safety, and higher patient compliance rates compared to standalone therepuetics.
Improving Device Functionality
Stents have been widely used since the 1990s. They offer many advantages compared to previous treatments. Unfortunately, patients treated with stents can develop stenosis, leading to complications and blood vessel occlusion.
In 2003 the FDA approved the first drug-eluting stent. A drug-eluting stent combines a drug enclosed in a polymer that coats the metallic framework. While drug-eluting stents add complexity and cost, they consistently outperform their standalone counterparts.
Combination Devices: Improving Therapeutic Effect and Patient Compliance
Transdermal patches provided a novel method for controlled, subcutaneous delivery into the bloodstream. They offered an alternative to invasive procedures using needles or infusions. Additionally, transdermal patches allowed gradual absorption of drugs which proved too potent to deliver orally without off-target effects.
Combination Products & Regulatory Requirements
Despite their advantages, combination products present new technological and organizational challenges. They require new product development strategies compared to standalone products, drugs, and devices.
Changes In The US: Good Manufacturing Practices and the ‘Streamlined Approach’:
cGMPs are crucial across the pharmaceutical industry. Unfortunately, the practices for drug devices and medicinal products have developed separately, and very few people are familiar with both. Combination products need to adhere to two separate rulesets, which has created a great deal of confusion.
In the US, these rules apply whether the product is sold as a single package or separately as a ‘referenced’ or ‘cross-labelled’ combination. The FDA’s final guidance document issued in 2015 states that “The cGMP requirements for constituent parts of cross-labelled combination products that are entirely manufactured at separate facilities are the same as those that would apply if these constituent parts were not part of a combination product.”
Despite this, there have been attempts at simplifying the process. An alternative ‘streamlined approach’ has been developed to identify cGMP regulations that would not be met by complying with device QS regulations and vice versa. This approach highlighted eight examples of drug cGMP regulations that would not be met by complying with device QS regulations and six examples of the device QS regulations that would not be met by complying with the drug cGMP regulations.
Theoretically, pharmaceutical companies can their drugs approved simply by adding an understanding of these additional rules to their pre-existing knowledge.
Changes in Europe: Article 117 of European medical device regulation
In the EU, the requirements for bringing combination products have recently undergone significant changes with the introduction of Article 117. Speaking at Formulation and Delivery 2021, Fatima Bennai-Sanfourche, Senior Manager at Bayer, explains that “this article is impacting combination products and getting European Medicines Agency and the European Comissionto work together to find a way to control this type of product. This is especially relevant to integral drug-device combinations.”
These changes present a challenge for pharmaceutical companies. They will have to rethink their current approaches to comply with the drug and device regulations during the development of drug-device combination products. Article 117 requires manufacturers of combination products to demonstrate that the device constituent conforms to the General Safety and Performance Requirements (GSPRs) of Annex I of the MDR.
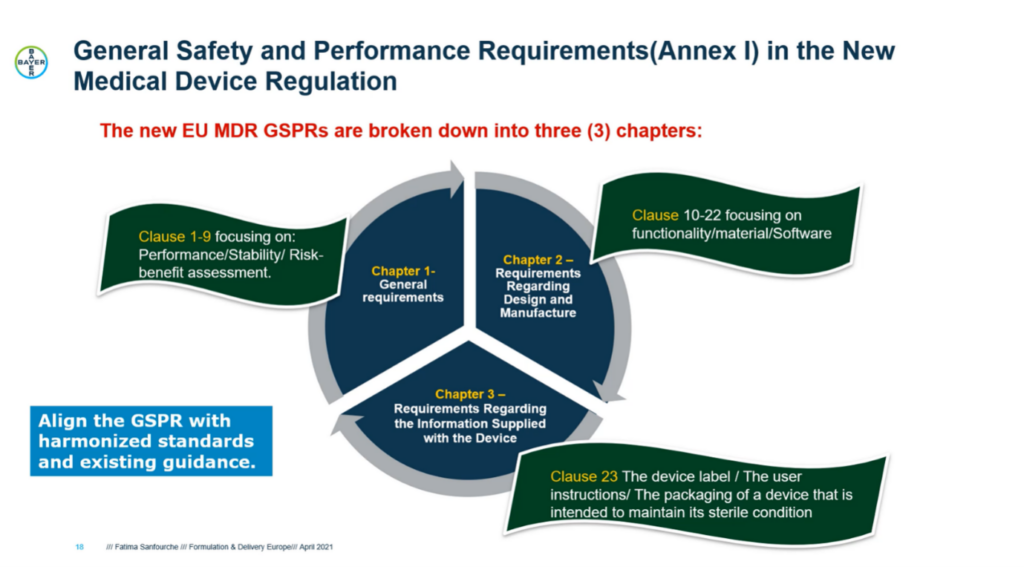
Annex I is split into three chapters. The first chapter details general requirements, focusing on performance, stability, and risk-benefit assessment. Fatima explains that “the first step is to create a risk management plan and implement risk mitigation. Once you do that, you need to identify any additional measures for enhancing the safety profile, for example, using alarms. We also need to ensure communication of the safety information to the patient.”
The second chapter centres on functionality, design and manufacturing requirements, while chapter three is concerned chiefly with device labelling. All the information on the device, packaging, and labelling must comply with the new rules.
Final Thoughts and Conclusion
Combination products have proven their worth, enabling new and improved therapeutic effects and increasing medication efficacy. The reason why drug-device combinations are so appealing is that they decrease the burden of taking medication. Additionally, these products decrease adverse effects because they reduce the likelihood of overdosing or underdosing on a prescribed medication. Unfortunately, defining what is and what isn’t a combination product is becoming increasingly difficult. Regulatory and legal bodies are working to provide clarity, such as the MDCG 2022-5 guidance from the European Commission published in 2022.
For more on combination products, consider attending our upcoming Formulation & Delivery: UK Event. Across two days, this event will feature over 50 technical presentations and case studies focused on the crucial issues in dosage form development, developing novel therapeutics, drug delivery devices, and combination products.
Related Resources