




.png)
Artificial Intelligence for Drug Design and Discovery

It is becoming ever more apparent that we will begin to see artificial intelligence radically transform our lives in the coming years and decades. We are just on the cusp of using machine learning as a tool to solve problems big and small while increasing efficiency and decreasing human error.
Moderated by Dr Sree Vadlamudi, Senior Director and Head of Business Development EU at Iktos, Oxford Global hosted an absorbing panel discussion on artificial intelligence applications for drug design and discovery. On the panel were Dr Eric Martin, Director of Computational Chemistry at Novartis, and Dr Leena Otsomaa, Vice President of Medicine Design and R&D at Orion Corporation.
Optimising the DMTA Cycle with Artificial Intelligence and Automation Methods
A breakthrough way in which artificial intelligence can be used effectively in the drug discovery process is in shortening the design-make-test-analyse (DMTA) cycle. Although this is typically expected to take up to four months, Vadlamudi predicted that AI could reduce this time by a factor of four; down to one month.
The vector from HIT finding to preclinical candidate nomination requires many DMTA cycles as any project progresses. These cycles are a major source sluggishness in the drug discovery process, so there has been interest in optimising them to speed up the discovery of new medicines.
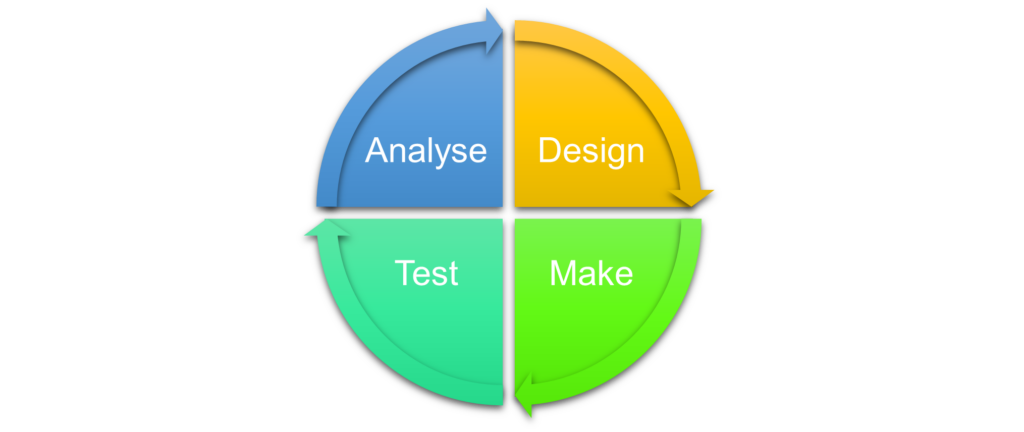
So how is artificial intelligence promising to improve the DMTA cycle? Vadlamudi points out that “everyone involved in the drug discovery process will ask: What should be the next molecule we create to progress the project to the next stage of discovery?” And that's where he says generative AI can help design novel molecules.
Here, Vadlamudi explained how Iktos’s Makya platform provides optimal in-silico easy to make molecule generation. Combining generative design with reinforcement learning, Makya can turn project data into in silico leads which can then be reviewed and analysed on the platform.
Vadlamudi says this technology is "of course, only the beginning of this process, but if one could couple these tools with automation, they could sharpen the goal of cutting down the DMTA cycle from three to four, to one month.”
"Massively Multitask Machine Learning Models"
Massively multitask machine-learning models are a way of sharing machine-learning across thousands of assays. This means that machine-learning algorithms have a pool of chemical structures and bioactivity measurements thousands of times larger to make their predictions.
“This dramatically improves the scope and accuracy of the predictions,” said Dr Eric Martin, CADD Director of Novartis. Martin builds massively multitask Profile-QSAR (pQSAR) models for 13,000 assays. Those models cover 2 million compounds and 20 million IC50s, updated every month.
Martin says that the models he builds have an average accuracy “comparable to 4-concentration dose-response experiments.” However, he claims that the applicability domain could be expanded further still, as it is currently limited to compounds similar to those in Novartis’s historical archive.
Martin explains that “if pharmaceutical companies would share their databases of chemical structures, assays, and bioactivity data, ‘super-massive’ multitask models would cover the chemical space of their combined collections.” But Martin explained that “of course,” they were reluctant to do so.
Collaborative pQSAR is Martin’s method of sharing this data naturally and safely; he says, “without really sharing it.” Profile-QSAR consists of two levels of stacked models. The conventional level-1 single-task models create a profile of bioactivity predictions. These profiles are then used as the compound descriptors for the level-2 pQSAR models.
This means that companies can safely share their level-1 models in a compiled format that Martin likens to “black-boxes”. These shared level-1 models can be used for an expanded profile for companies to generate their own level-2 models. “The result is mathematically identical to the pQSAR models they would have built if they had actually shared the experimental data,” claimed Martin.
Martin could simulate this kind of collaboration because Novartis was formed from the mergers of several smaller companies. Disappointingly, it did not improve predictions for each company’s internal compounds, but it did for the collaborator’s compounds. The simulations also showed that increased overlap between the partners’ compound collections increased the benefit of collaboration.
While the method was shown to work, practical challenges remain. Companies that contribute few level-1 models benefit significantly more than companies that contribute many. Martin suggested that this could be resolved by capping the number of level-1 models a company could receive from each partner to the same number they contribute.
Martin explained that this capping, however, might encourage companies to “cheat” by contributing bogus models or models trained on public datasets. So, an auditor might be needed to ensure each company had the experimental data to support their contributed level-1 models.
Predictions for the Influence of Artificial Intelligence on the Future of Medicinal Chemistry
Dr Leena Otsomaa declared at the beginning of her part of the discussion that “we don’t have AI yet.” Explaining that what we instead had was machine learning methods that are helping us improve our models. The difference here was that scientists are not yet at a stage where they can claim that their algorithms produce entirely new models outside of existing data points.
The problem with the way AI is implemented today is that companies that provide AI solutions and drug discovery companies are detached from each other. But Otsomaa says that some start-ups are taking a different innovative approach. Therefore, Otsomaa stated that there has to be a close connection between the development of AI and other people in science and tech, “they have to be intertwined.”
Otsomaa said that she “truly believed” that AI will have a comparable impact to the discovery of electricity, “it started as a power source to industry and then made its way into the home, stretching out into all parts of everyday life. She predicted that we would eventually see revolutionary breakthroughs using AI, but we’re not there yet, and we can only begin to speculate what kind of applications these may be.
How fast will the development time be for AI compared to the 200 years it took to develop electricity to where it is today? "I believe that it will be faster,” Otsomaa claimed, “but to ensure that there is a fruitful development of AI, I believe that there ought to be a mix between people from different disciplines.” She explained that if this is not the case, we lose the possibility of identifying where AI could most optimally be used.
We are not yet at the point where we are using active learning as part of the process, and when this happens, Otsomaa predicts it will be a “paradigm shift.” Otsomaa ended her presentation with a prediction as to what will be required from scientists when AI becomes more ever-present:
“We are now synthesising drug compounds with a view towards them becoming the drug candidates, but it's not yet in our daily thinking to create compounds to improve the model. This will definitely impact competencies—what is required from chemists in drug discovery. Chemists will need to begin to develop a better understanding of data science as well as synthetic chemistry. Importance of data science understanding applies to other scientists in the field of drug discovery as well.”
Machine learning is a topic that we often cover on our online content portals, so to track the latest updates in how artificial intelligence is being used in Drug Discovery, please sign up to our monthly newsletters. To see the experts in the field in-person, consider joining us at our upcoming Discovery Europe conference.

